
Regulatory Problems about Biomaterials and Medical Devices

Guidelines for post-market surveillance of medical devices
March 2, 2022In brief
All biomaterials and medical devices must comply with various Pharmaceutical Regulatory standards and rules to receive clearance. It covers a range of processes and regulations, including commercialization, clinical development, good manufacturing practice, and post-market surveillance. Global rules and regulatory challenges relating to biomaterials and medical devices are discussed. Particularly effective for smaller businesses that may not have the funds to hire a full-time vigilance specialist. Risk management, intellectual property protection, marketing authorization, university patent licences, and general good manufacturing practice are covered in the methods and regulations.
Introduction
Medical Device Regulatory Affairs has many advantages for patients, but they also have a lot of risks, especially those meant to be implanted. The purpose of national medical device regulatory authorities is to safeguard patients and the general public from dangerous goods while also allowing for the rapid introduction of novel technologies that can benefit patients and promote public health.
Based on risk assessment, the regulatory framework and standardized test techniques for medical devices, including biomaterials, are now harmonized worldwide. In this broad context, new implanted biomaterials should pass a set of regulatory tests before receiving marketing authorization. Physical and chemical categorizations, biological in vitro and in vivo studies, and clinical investigations require examinations. For traditional biomaterials and implanted devices, this approach is well-established. However, emerging revolutionary goods, such as Nano biomaterials, tissue engineering materials, and 3D-bioprinting tissues and organs, confront regulatory difficulties that act as hurdles to their timely delivery to patients.
The most recent research on the contradictory concerns emerges when evaluating newly created biomaterials and medical devices. First, a background on regulatory procedures in the United States and the European Union is presented, and a discussion of the definition of medical innovation in the medical device regulatory consulting services. The links between medical device legislation and innovation are examined on this basis. Finally, examples of cutting-edge medical technology are examined.
Methods
This open search corrected other original scientific publications and reports relevant to the articles studied in the systematic search from various regulatory bodies, consulting and expert groups, and governmental departments. These publications are especially significant because numerous stakeholders are concerned about the ethical, scientific, and legal aspects of medical device innovation, so these topics are covered in several places. However, not all of the publications evaluated in our study are cited owing to length limitations.
Medical device regulations
The Device Amendments of the United States Food and Drug Administration (FDA) produced the first regulation for devices intended for human use, and the FDA had a unique position among national regulatory bodies all over the globe until recently. The Center for Devices experts in regulatory affairs and Radiological Health (CDRH) is now in charge of premarket approval of medical devices in the United States and post-market surveillance of these devices.
Most Class I medical devices must be registered with the FDA but are not required to undergo a 510(k) premarket review. However, they must adhere to general controls such as manufacturer registration and announcement to the FDA before marketing, good manufacturing performs (GMP), appropriate branding and labelling, and available post-market reporting procedures. To meet regulatory criteria and post-market supervision, Class II medical devices must have particular labelling requirements.
Clinical studies, done in line with Good Clinical Practices (GCP), are required by the FDA for “high-risk” devices to establish safety and effectiveness. These trials can be costly and take years to complete. After receiving FDA investigational device exemption (IDE) approval, the devices are clinically approved. Unless its “substantially equivalence” to a lawfully sold Class I or Class II item can be proven, a device that has not been on the market is deemed a Class III.
Regulations’ Impact on Medical Device Innovation
Regulatory concerns influence the whole innovation cycle, and they must be considered throughout the design and development of medical devices and during pre-clinical and clinical testing, product regulatory testing, manufacturing, and post-marketing surveillance. As a result, the collaboration between medical device developers and national regulatory bodies is crucial for innovation and competitiveness.
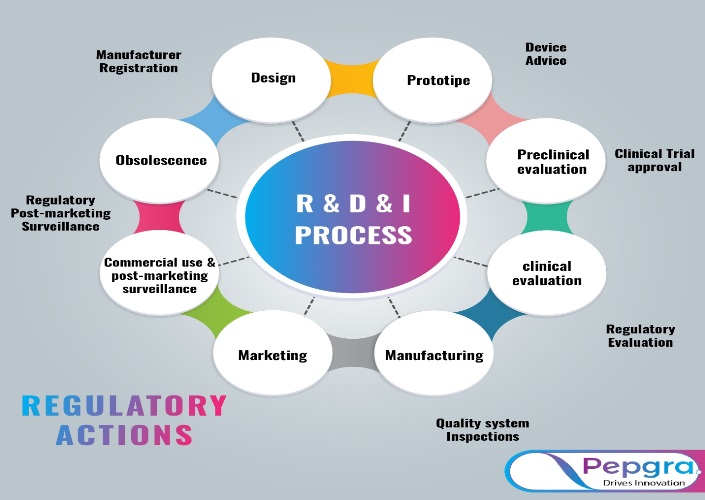
To show compliance with the “Essential Principles of Safety and Performance of Medical Devices,” quality and risk management systems must be incorporated throughout the life cycle of medical goods. The identification and description of dangers connected with medical devices and their accessories; risk analysis, evaluation, and control; and monitoring the efficacy of that control are all part of risk management.
Conclusion
Strong medical device laws exist to safeguard patients and the rest of society from dangerous items and improve public health by bringing new products to market that can benefit patients. However, the risk of releasing devices that haven’t been thoroughly tested exists, and an effective review procedure is still a source of worry for academics, business, government, and the general public. The categorization of medical goods into pharmaceutical regulatory affairs, biologics, and medical devices is the cornerstone of the world’s most advanced regulatory systems; nevertheless, novel combination products prove problematic to the present product classification scheme. Furthermore, the use of emerging technologies in medical device design, such as nanotechnology, tissue engineering, and 3D printing, emphasizes classifying medical devices into risk classes. Even at the earliest phases of the invention process, the duty for ensuring the safety and efficacy of devices relies not only on national regulatory authorities but largely on researchers, manufacturers, and clinicians.
About Pepgra
Pepgra CRO provides high-quality Clinical and Regulatory Expert Services to its clients. Our regulatory services experience draws heavily on the most recent research to assist you in designing and implementing clinical and regulatory frameworks that meet your needs. Our pharmaceutical and biotechnology industries clients have been very pleased with our regulatory services. Our regulatory affairs professionals from the world’s leading pharmaceutical companies focus on assisting in developing new medical products, the integration of regulatory principles, and authoring and submitting appropriate reports to health authorities.
References
- Guerra-Bretaña, Rosa Mayelin, and Andrea Lucía Flórez-Rendón. “Impact of regulations on innovation in the field of medical devices.” Research on biomedical engineering34 (2018): 356-367.
- Gurman, Pablo, O. Rabinovitz, and Tim B. Hunter. “Regulatory challenges on biomaterials: focus on medical devices.” Biomater Sci Integr Clin Eng Approach223 (2012): 48.

