
Literature Screening and Risk Management for Biosimilars – Challenges and Preventive Measures

Uses and Implementation process of Electronic Data Capture (EDC) in Clinical Trials
February 23, 2021
Evaluating the results of the literature search and screening
February 28, 2022In-Brief
- Biosimilars signify a new class of medical products that will significantly impact the clinical practice of pharmacovigilance literature search.
- They are the same on an amino acid sequence level to present reference biopharmaceutical products. However, they may show differences on a protein level.
- Pepgra blog provides a brief overview of biosimilar development. It describes the preventive measures and challenges that should be considered during bio similars’ admission into the clinic using literature surveillance in pharmacovigilance and provides pharmacovigilance literature search services.
Introduction
The US FDA characterizes a biosimilar as “a natural product that is profoundly like a US-authorized reference organic product despite minor contrasts in clinically idle parts. For which there are no clinically significant contrasts between the organic product and the reference product regarding the security, virtue, and intensity of the product.” The European Medicine Agency definition is practically identical in literature screening.
The World Health Organization characterizes pharmacovigilance as the science and exercises identifying with the recognition, evaluation, comprehension, and anticipation of unfavourable impacts or other medication-related issues.
Assembling the two has yielded a complex administrative scene with wide varieties and irregularities across nations and markets.
What’s reasonable is that it is adequately troublesome to assemble and keep a powerful PV program to meet administrative prerequisites for little particle drugs – but such projects won’t fulfil the necessities for biosimilars.
Organizations creating biosimilars, regardless of whether from their trendsetter biologic or another that has gone off-patent, should know about a few significant questions as for biosimilars that will affect their pharmacovigilance literature screening services.
Challenges in Literature screening and risk management for biosimilars:
Assembling strategies
The assembling cycle for biopharmaceuticals is more perplexing than for conventional little particle drugs. Little contrasts between assembling strategies can altogether affect a biosimilar’s natural properties, perfection and clinical action. Consequently, there is no assurance that the subsequent biosimilar will be equivalent to its reference product.
Product names
Fifty particular biosimilars are presently being developed—but since their names are not unmistakable, this groundswell is probably going to bring about visibility issues in case of an ADR, at any rate temporarily. That is because biosimilars in the EU can have a similar International Non-proprietary Name (INN) as the trendsetter biologic. The FDA plans to assign a non-proprietary name that incorporates a postfix made out of four lowercase letters. Nonetheless, in reality, patients and clinicians may keep on alluding to a biosimilar by its reference image name, even in ADRs from scientific literature search services.
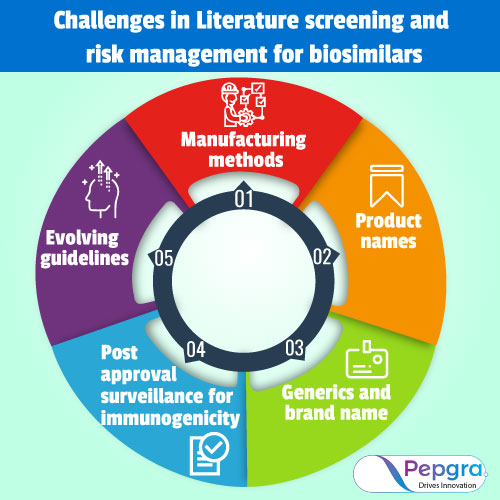
Generics and brand name
Products can be recommended reciprocally by and large. Biosimilars—albeit tantamount to the trendsetter medications—can’t. “Programmed” compatibility would require information showing that a biosimilar produces an identical clinical outcome in some random person. The FDA still can’t uncover how it will deal with biosimilars’ compatibility, though the EMA leaves the individual party states’ choice.
Post endorsement observation for immunogenicity
Post endorsement observation for immunogenicity and uncommon unfavourable occasions might be required or potentially needed over the long-term when a biosimilar is available. Such observing is commanded in the EU, although the FDA presently can’t explicitly address this issue.
Advancing rules
As rules for biosimilar endorsements and PV advance, particularly in the US, drug organizations should remain cautious with the goal that their PV projects can quickly and effectively adjust to developing administrative measures.
Preventive measures
- Maintaining a vault of data on natural products accessible in the district will help in right recognizable proof of the product elaborate when a negative response is accounted for in the PV framework.
- Developing unique contents that would take into consideration the assortment of point by point data of the product connected with the unfriendly response in the underlying or follow up correspondence
- Ensuring cautious clinical assessment of all speculated immunogenicity reports with comprehension
- Implementing continuous total survey of wellbeing information and examination with the security profile of the reference product to comprehend the distinctions in danger profiles
- Designing an RMP with extra measures to distinguish or assess obscure security issues, including immunogenicity and uncommon occasions, yet
- Setting up unique product/tolerant vaults for associate occasion checking
- Conducting controlled post-endorsement adequacy and wellbeing concentrates signs and target populaces altogether sufficiently
- A product name with viability and security data was identified with both the reference product and biosimilar recognized by source.
Conclusion
End-of-patent exclusiveness and developments in biotechnology, enabling their producers, have created substantial opportunities for follow-on biologics or Biosimilars to arrive at the market and serve patients’ requirements all over the world in a cost-effective manner. However, Biosimilars’ PV and risk management present many unique and special challenges. Pepgra also listed the preventive measures to control the risks in clinical sectors and provides pharmacovigilance literature screening services.
References
- Casadevall, N., Edwards, I. R., Felix, T., Graze, P. R., Litten, J. B., Strober, B. E., & Warnock, D. G. (2013). Pharmacovigilance and biosimilars: considerations, needs and challenges. Expert Opinion on Biological Therapy, 13(7), 1039-1047.
- Scavone, C., Rafaniello, C., Berrino, L., Rossi, F., & Capuano, A. (2017). Strengths, weaknesses and future challenges of biosimilars’ development. An opinion on how to improve the knowledge and use of biosimilars in clinical practice. Pharmacological research, 126, 138-142.
- Zuñiga, L., & Calvo, B. (2010). Biosimilars: pharmacovigilance and risk management. Pharmacoepidemiology and drug safety, 19(7), 661-669.
